|
Getting your Trinity Audio player ready... |
R&D Guide to Dissolution Specification Setting- Global regulatory
Dissolution Specification Setting: Establishing dissolution specifications during the research and development (R&D) stage is one of the most critical activities in pharmaceutical product development. These specifications act as a bridge between laboratory findings and real-world drug performance, ensuring that the formulation consistently delivers therapeutic benefits while also meeting global regulatory requirements.
Dissolution testing is not simply a quality control check; it is a predictive tool that provides insight into how a drug is likely to behave in the human body. A well-defined dissolution specification reduces the risk of failure during clinical evaluation, streamlines regulatory submissions, and ensures consistent product quality throughout the product’s lifecycle.
This guide provides a structured, step-by-step approach to setting dissolution specifications in R&D, supported by regulatory considerations and scientific rationale.
1. Understand the Role of Dissolution: Dissolution Specification Setting
Dissolution testing measures how quickly and to what extent a drug substance is released from its dosage form into solution. For oral solid dosage forms—such as tablets, capsules, or modified-release formulations—this step is crucial, since drug absorption is largely dependent on dissolution rate.
A slow or incomplete dissolution could compromise bioavailability, leading to therapeutic failure, while overly rapid dissolution might alter drug exposure or increase side effects. By defining dissolution specifications early, developers can anticipate and control variability, ensuring that the drug will behave predictably in vivo.
2. Benchmark Against the Reference Product: Dissolution Specification Setting
For generic drug development, the starting point is always the Reference Listed Drug (RLD), or the innovator formulation. The reference product serves as the standard for evaluating whether the new formulation is equivalent in terms of performance.
The key steps include:
- Comparative Dissolution Testing: Conduct studies using multiple physiologically relevant media, typically pH 1.2 (gastric), pH 4.5 (intestinal), and pH 6.8 (buffer).
- Discriminatory Power: Identify the medium that best differentiates between formulations, as this will help in selecting a sensitive method.
- Similarity Assessment: Compare profiles using the f₂ similarity factor, where an f₂ value of ≥ 50 indicates that the profiles are sufficiently alike.
This approach ensures that the test product demonstrates dissolution performance comparable to the innovator under relevant physiological conditions.
3. Apply Biopharmaceutics Classification System (BCS) Principles: Dissolution Specification Setting
The Biopharmaceutics Classification System (BCS) is a critical framework when setting dissolution specifications. It categorizes drugs into four classes based on solubility and permeability, and these categories determine how sensitive a formulation is to dissolution changes:
- BCS Class I (High Solubility, High Permeability): Dissolution is usually not a limiting factor for absorption. Specifications can be broader, as in vivo performance is less dissolution-sensitive.
- BCS Class II (Low Solubility, High Permeability): Dissolution is often the rate-limiting step for absorption. Tight dissolution specifications are essential, as variability could directly affect bioavailability.
- BCS Class III (High Solubility, Low Permeability): Absorption is limited by permeability rather than dissolution. Specifications still matter but can be somewhat less stringent.
- BCS Class IV (Low Solubility, Low Permeability): Both solubility and permeability pose challenges. Dissolution specifications must be carefully justified, often with additional data.
By applying BCS principles, developers can balance clinical relevance with practicality, ensuring specifications are neither too restrictive nor too lenient.
4. Design a Discriminatory Dissolution Method: Dissolution Specification Setting
A robust dissolution method should be discriminatory, meaning it can detect changes in critical quality attributes (CQAs) of the product, including formulation composition, process variations, and active pharmaceutical ingredient (API) properties.
Key considerations include:
- Selection of Medium: Choose dissolution media that reflect physiological conditions and highlight differences between batches if present. For poorly soluble drugs, surfactants such as sodium lauryl sulfate (SLS) may be added to improve discrimination.
- Choice of Apparatus: Use an apparatus (e.g., USP Apparatus I – basket, or Apparatus II – paddle) that best mimics the intended in vivo conditions. For modified-release products, specialized apparatus may be required.
- Method Robustness: Ensure the method is reproducible and sensitive enough to detect variations in formulation or process.
Designing a discriminatory method early prevents issues later during scale-up and regulatory submission.
5. Establish a Tentative Specification: Dissolution Specification Setting
Once comparative studies and method development are complete, the next step is to propose a tentative dissolution specification. This is usually expressed in terms of Q, the percentage of drug dissolved at a specified time.
Example: Immediate-Release (IR) Tablets
A typical format for IR products is:
- Not less than (NLT) 80% of labeled drug content dissolved within 30 minutes.
However, this specification must be adjusted based on:
- The behavior of the RLD.
- Bioequivalence (BE) risk.
- Clinical relevance of the dissolution rate.
Example: Modified/Extended-Release (MR/ER) Products
For MR or ER formulations, specifications often require multiple time-point targets to ensure sustained release:
- 20–40% in 1 hour,
- 50–70% in 4 hours,
- NLT 80% in 12 hours.
These staged criteria ensure that the release profile is consistent with therapeutic needs and mirrors reference product behavior.
6. Refine After Bioequivalence Studies
The tentative specifications set during R&D are later refined after BE studies. Once bioequivalence is demonstrated in clinical trials, dissolution specifications should be adjusted to reflect the actual release characteristics of the batches used in BE studies.
At this stage, specifications transition from tentative to final commercial specifications, which are included in regulatory filings and become the basis for quality control during production.
7. Align with Regulatory Expectations
Regulatory agencies worldwide provide guidance on dissolution testing and specification setting. Developers must align their approach with the expectations of the target market.
- USFDA (United States Food and Drug Administration): Provides guidance on dissolution testing, scale-up and post-approval changes (SUPAC), and BCS-based biowaivers.
- EMA (European Medicines Agency): Issues guidance on bioequivalence and dissolution testing, emphasizing discriminatory methods and similarity assessment.
- WHO (World Health Organization): Offers requirements for multisource (generic) products, focusing on stability and quality control dissolution testing.
Understanding and following these frameworks ensures smoother regulatory review and global acceptance.
Best Practice Tip: Justify Specifications Scientifically
The most important principle in setting dissolution specifications is scientific justification. Specifications should not be arbitrary but grounded in:
- Comparative Data: Reference product performance and f₂ similarity analysis.
- Risk Assessment: Understanding of how formulation and process variables affect dissolution.
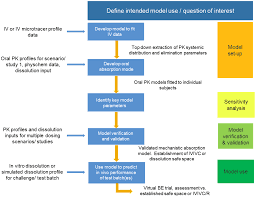
- In Vitro–In Vivo Correlation (IVIVC): If available, IVIVC data strengthens the case that dissolution results predict clinical performance.
A well-justified dissolution specification enhances regulatory confidence, facilitates approval, and ensures patient safety.
Conclusion
Dissolution specification setting is both a science and an art. At the R&D stage, it requires careful consideration of reference product behavior, BCS classification, method development, and clinical relevance. Tentative specifications should be flexible enough to guide development yet rigorous enough to ensure performance.
As the product advances through bioequivalence studies and regulatory submissions, these specifications are refined into final criteria that safeguard consistency and quality in commercial manufacturing.
By combining comparative data, regulatory guidance, and scientific reasoning, developers can set dissolution specifications that not only meet compliance requirements but also ensure reliable therapeutic performance for patients worldwide.