|
Getting your Trinity Audio player ready... |
Q4B Annex 11 Capillary Electrophoresis General Chapter
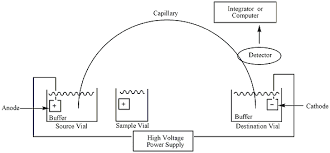
1. Introduction: Capillary Electrophoresis
Capillary Electrophoresis : This annex results from the ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use) Q4B process, focusing on the General Chapter for Capillary Electrophoresis. The proposed guidelines and texts were submitted by the Pharmacopoeial Discussion Group (PDG). This annex offers a harmonized approach for the implementation of Capillary Electrophoresis methods within the regulatory frameworks of different regions, aiming to align and standardize practices across the globe. It is a part of ongoing efforts to create consistency in pharmaceutical practices, ensuring that methods and standards are both accurate and reliable for the global market.
2. Q4B Outcome
2.1 Analytical Procedures
Following an extensive evaluation by the Q4B Expert Working Group (EWG), the ICH Steering Committee has concluded that the analytical methods described in the official pharmacopoeial texts of various regions can be used interchangeably. These texts include:
- Ph. Eur. 2.2.47: Capillary Electrophoresis (European Pharmacopoeia).
- JP General Information 4: Capillary Electrophoresis (Japanese Pharmacopoeia).
- USP General Information Chapter <1053>: Biotechnology-derived Articles – Capillary Electrophoresis (United States Pharmacopeia).
The ICH has confirmed that the contents of these chapters from the respective pharmacopoeias can be accepted as interchangeable for use across the ICH regions. This harmonization aims to facilitate smoother regulatory processes and reduce the complexity involved in the approval of pharmaceutical products internationally.
2.2 Acceptance Criteria
The reviewed texts did not include specific acceptance criteria, which may imply that regional authorities or the involved stakeholders will have to define acceptance criteria based on their specific needs or regulatory requirements. This provides flexibility while maintaining compliance with global standards for pharmaceutical testing.
3. Timing of Annex Implementation
The implementation of this annex will occur when it is formally incorporated into the regulatory process under ICH Step 5. It is important to note that the timing of this incorporation will differ across various regions. Some regions may adopt the new guidance sooner than others, and local regulatory bodies will determine how quickly they incorporate the guidance into their pharmaceutical and analytical practices.
- Considerations for Implementation
The adoption of the harmonized guidelines presented in this annex requires careful consideration, as pharmaceutical companies and manufacturers may need to adjust their existing methods. The following points are crucial to consider in different regions:
4.1 General Considerations
Pharmaceutical sponsors or manufacturers wishing to change their existing methods to align with the new pharmacopoeial standards referenced in Section 2.1 of this annex must adhere to the regulatory mechanisms of the respective regions. These procedures may involve submitting change notifications, variation requests, and/or obtaining prior approval from the relevant regulatory authorities. Such notifications must follow established protocols designed for compendial changes, ensuring that the methods remain scientifically valid and regulatory compliant.
4.2 FDA Considerations
In the United States, the Food and Drug Administration (FDA) is responsible for ensuring that pharmaceutical companies implement the standards outlined in this annex. While the FDA has agreed that the referenced pharmacopoeial texts are interchangeable, they may still require companies to demonstrate the suitability and acceptability of their chosen method for specific materials or products. This means that the FDA may ask for additional evidence or data to verify that the adopted method performs appropriately, even if it is sourced from a different pharmacopoeia. Therefore, while the interchangeability has been recognized, the FDA still emphasizes the need for method validation based on product-specific requirements.
4.3 EU Considerations
For the European Union, the regulatory authorities will accept the reference to the pharmacopoeial texts as specified in Section 2.1 when a marketing authorization application, renewal, or variation is submitted. This allows applicants to cite the appropriate text from another pharmacopoeia as per the terms outlined in this annex. This flexibility ensures that applicants can meet the requirements of the European Pharmacopoeia (Ph. Eur.) Chapter 2.2.47 on Capillary Electrophoresis by referencing equivalent texts from other established pharmacopoeias, as long as the interchangeability is declared and justified.
4.4 MHLW Considerations
The Ministry of Health, Labour, and Welfare (MHLW) in Japan also recognizes the interchangeability of the pharmacopoeial texts as referenced in this annex. However, Japan has specific implementation requirements that will be detailed when the annex is officially adopted by the MHLW. As with other regions, the MHLW will ensure that the new guidance is in line with Japanese pharmaceutical standards, offering clarity on how companies should incorporate the changes into their processes.
4.5 Health Canada Considerations
In Canada, the use of any of the referenced pharmacopoeial texts in accordance with the conditions set out in this annex will also be considered interchangeable. The Canadian regulatory authorities will accept these texts for compliance, as long as the adoption is in line with the established guidance provided in this annex. This harmonized approach simplifies the regulatory landscape in Canada, providing consistency with other global markets.
5. References Used for the Q4B Evaluation
The Q4B evaluation relied on several important references that shaped the outcome of this annex. The key documents are as follows:
5.1 PDG Stage 5B Sign-Off Document:
The PDG Stage 5B sign-off document, published in the Japanese Pharmacopoeial Forum, Volume 11, number 4 (October 2002), formed a critical part of the evaluation process. This document presented the final sign-off on the harmonization effort, reflecting the consensus reached by the international stakeholders.
5.2 Pharmacopoeial References for Capillary Electrophoresis General Chapter:
The following pharmacopoeial texts provided the basis for the Q4B evaluation:
- European Pharmacopoeia (Ph. Eur.): Supplement 6.6, published in June 2009 and officially implemented on January 1, 2010. The relevant text is identified as Capillary Electrophoresis (Reference 01/2008:20247).
- Japanese Pharmacopoeia (JP): General Information 4, as outlined in the JP Fifteenth Edition (March 31, 2006). The text was updated in May 2010 by errata published by the Ministry of Health, Labour, and Welfare (MHLW), which is accessible online via the Ministry’s website.
- United States Pharmacopeia (USP): Chapter <1053> – Biotechnology-derived Articles – Capillary Electrophoresis, which was first introduced via a USP Revision Bulletin on May 29, 2009, and became official on July 1, 2009. The text was a significant addition to the USP to align with the increasing importance of Capillary Electrophoresis in biotechnology-derived pharmaceutical products.
6. Conclusion
The Q4B Annex 11 provides a crucial step in the international harmonization of Capillary Electrophoresis methods used in pharmaceutical testing. By recognizing the interchangeability of pharmacopoeial texts from the European, Japanese, and United States Pharmacopoeias, the ICH aims to simplify the regulatory processes for companies that must meet global standards. However, despite the broad acceptance of these texts, each region’s regulatory authorities will continue to enforce their unique implementation requirements, ensuring that pharmaceutical companies validate and demonstrate the suitability of their chosen methods for each specific product. This collaborative effort supports the goal of improving pharmaceutical quality and safety while reducing the regulatory burden for global companies.