|
Getting your Trinity Audio player ready... |
Online f1 and f2 factor calculator in dissolution software free
Online f1 and f2 factor: A biowaiver refers to the exemption from conducting in vivo bioequivalence or bioavailability studies. Instead of relying on expensive and time-consuming bioequivalence trials, dissolution profile comparison can serve as a substitute for determining whether two drug formulations are equivalent. This review highlights the expectations of different regulatory authorities regarding biowaiver studies and their reliance on dissolution testing, particularly the model-independent similarity factor (f2) method.
Initially, regulatory agencies did not define the “early time point” in dissolution testing; however, the U.S. FDA has recently established this as within 10 minutes for products designed for rapid or immediate release. The Biopharmaceutics Classification System (BCS)-based biowaiver concept aims to minimize the need for in vivo studies by providing reliable in vitro alternatives. A waiver may be granted if adequate dissolution data justify the drug’s in vivo performance.
The BCS approach evaluates drug solubility, permeability, and physicochemical characteristics. Among available analytical tools, the similarity factor (f2) has been widely recommended by regulatory agencies because it is simple, reproducible, and provides an accepted threshold (f2 ≥ 50) for determining profile similarity. Nonetheless, variations exist among international guidelines in defining acceptance criteria, such as the number of time points, endpoint selection, coefficient of variation, and exemptions from f2 application. Hence, harmonized guidelines are essential to create a globally applicable framework for biowaiver submissions, particularly in the context of Abbreviated New Drug Applications (ANDA).
Keywords: Biowaiver, In vitro, Similarity factor, Dissolution testing, Biopharmaceutics Classification System (BCS), Bioequivalence.
Download free Online Software link below:
Dilution&ppm
Drug dissolution profile
F1_F2_Dissolution_Calculator
INTRODUCTION:Online f1 and f2 factor
Dissolution testing measures the rate and extent to which a drug substance dissolves under controlled conditions. Oral solid dosage forms are the most widely used due to their stability, safety, and cost-effectiveness. However, the bioavailability of these products largely depends on drug solubility, gastrointestinal permeability, and physiological conditions.
Given this, the dissolution profile plays a central role in establishing therapeutic equivalence between products. The World Health Organization (WHO) has also encouraged the use of generics, particularly in developing regions, to ensure affordability and accessibility. A generic substitute is deemed acceptable when it contains the same drug substance in the same strength, dosage form, and route of administration as the innovator, and also complies with quality, purity, and potency requirements.
The similarity factor (f2) method has emerged as a widely accepted statistical approach for comparing dissolution profiles. Regulatory bodies across the globe recommend its use, yet its application is not standardized internationally. A biowaiver, by definition, implies that in vivo studies may be replaced by in vitro dissolution testing, provided adequate justification is available. Dissolution profile comparisons using the similarity factor are therefore valuable tools for demonstrating equivalence under various test conditions.
METHODS:Online f1 and f2 factor
A validated method is essential for performing dissolution studies on both the innovator and test formulations. Typically, three dissolution media are recommended, each simulating different regions of the gastrointestinal tract:
- 0.1 N HCl or simulated gastric fluid (SGF) without enzymes
- pH 4.5 acetate buffer
- pH 6.8 phosphate buffer or simulated intestinal fluid (SIF) without enzymes
These media reflect physiologically relevant conditions for assessing drug release patterns. In cases where alternative media such as water are used, justification must be provided.
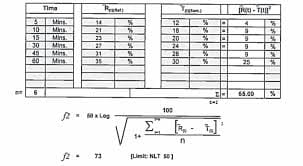
Moore and Flanner proposed two model-independent mathematical approaches to compare dissolution profiles: the dissimilarity factor (f1) and the similarity factor (f2).
- Dissimilarity factor (f1): Evaluates the percent difference between two profiles at each time point. A value closer to zero indicates greater similarity.
- Similarity factor (f2): A logarithmic reciprocal square root transformation of the mean squared differences, representing the degree of similarity. Values between 50 and 100 suggest equivalence.
According to regulatory standards, dissolution profiles of the reference and test products are considered similar when f1 < 15 and f2 ≥ 50. Generally, a minimum of 12 dosage units and at least three time points (excluding zero) are required for comparison. Variability limits are typically set at 20% for early time points and 10% for subsequent ones.
REGULATORY CONSIDERATIONS:Online f1 and f2 factor
Different regulatory agencies have issued guidelines for dissolution profile comparison, though notable discrepancies exist. Key highlights are as follows:
- United States (FDA): Requires at least three time points, with f2 ≥ 50. If >85% of the drug is released within 15 minutes, f2 calculation may be waived.
- Europe (EMA): Similar criteria as the U.S., with emphasis on reaching 85% dissolution or the asymptote.
- Japan: Provides detailed conditions, requiring three to eight time points depending on release rates in various media.
- Canada: Mandates sufficient time points until 90% drug release is achieved, with similar variability limits and f2 requirements as the U.S.
- Australia: Requires at least three time points, excluding zero, with the same f2 acceptance range.
- Brazil: Specifies a minimum of five time points, with variability and f2 criteria similar to other regions.
- India: Requires adequate time points for complete dissolution but does not clearly define f2 exemptions.
These regional differences highlight the lack of global uniformity in applying f2-based dissolution comparisons.
CONCLUSION
Comparison of international guidelines reveals significant variations in the application of dissolution testing and similarity factor evaluation. Differences exist in required time points, endpoint selection, coefficient of variation thresholds, and conditions for f2 exemption. The FDA’s recommendation of an early time point within 10 minutes for immediate-release products is not universally adopted, further contributing to inconsistencies.
To streamline the regulatory process, there is an urgent need for harmonized biowaiver guidelines that can be applied globally. Such alignment would simplify the filing process for ANDA submissions by enabling formulation developers to generate a single data set acceptable across multiple jurisdictions. Overall, the biowaiver approach, grounded in BCS and dissolution profile comparisons, offers a cost-effective and time-efficient alternative to traditional bioequivalence studies.